CDK Plotting Basics: Rotation and Center-Translation
tl;dr plotting molecules is fun once you get the hang of it.
This post is a continuation of my previous post, wherein I began playing with the Chemistry Development Kit (CDK) for the purpose of making visualizations. CDK does a great job of generating structures of small molecules, but what if you want to scale, transform, or rotate these molecule on your canvas? Its not quite as easy as it seems.
Let me show you basic depiction generation using Clojure. There are a lot of steps that require understanding at least a little bit of how Java renders although to be honest I am still trying to figure out the process. But by using several online resources for inspiration, I was able to get something working. So I can play around even if i don’t know why all of these generators and visitors are needed. Heres a basic renderer
(defn basicrender [mol]
(let [font (Font. "Verdana" Font/PLAIN 18)
;; make the generators that will set up and draw stuff
gen (doto (ArrayList.)
(.add (new BasicSceneGenerator))
(.add (new StandardGenerator font)))
;; create the Buffered image the molecule will get drawn to
image (BufferedImage. 800 300 1)
;; create a drawing area. "drawArea" is actually a misnomer.
;; This will not get drawn; it will only be used to
;; scale the molecule to an area
drawArea (Rectangle. 800 300)
g (doto (.getGraphics image) (.fillRect 0 0 800 300))
;; the renderer is where you can set stuff
renderer (AtomContainerRenderer. gen (new AWTFontManager))]
;; setup function. whats this do? its what scales the molecule
(-> renderer (. setup mol drawArea))
;; and finally paint. Paint will use the bounds obtained in the setup
;; and use this info to paint the molecule to `g`,
;; the graphics context of your image
(-> renderer (. paint mol (new AWTDrawVisitor g)))
image))
So this basic function works to generate an image; great. And I can use the GeometryTools/rotate function to rotate the molecule and it will rotate, so that is also great. But what about scaling? it won’t work (see below). Whats going on?
(def caffeine (parsesmiles "CN1C=NC2=C1C(=O)N(C(=O)N2C)C"))
(show (basicrender caffeine)) ;works!
(GeometryTools/rotate caffeine (Point2d. 100 0) 90 ) ;rotate the molecule 90 deg
(show (basicrender caffeine)) ;rotated 90!
(GeometryTools/scaleMolecule caffeine 0.5) ; not scaled! whatsup?
So the scaling/translation is not going to work. Something is happening during the conversion from molecule units to screen units that is undoing the scaling/transformation. A quick perusal of the mailing list comes up with a nice overview of the issue:
…To give a concrete example, lets say we have H2 in a molfile. The distance between the atoms is 1 unit (not necessarily a real unit, like picometers). If the bond length is 10 pixels, then the scale is
- If the zoom is then 0.9, then the line drawn to represent the bond will be 9 pixels.
The reason for this is that models may have different scales, and the transform uses this to render at a particular bond length. The zoom is user-settable, and only reflects how to change the bond length. So really, it is bond length and zoom that are redundant - they are different ways to think of the same thing, specifically how long the lines are between atoms…..
Okay, so my basic suspicions were confirmed but I still don’t know what to do. Although this gives me an idea of the problems the developers faced I am still uncertain how to scale and transform. Lets look at the .setup() function in the AtomContainerRenderer class that is called during molecule generation.
public void setup(IAtomContainer atomContainer, Rectangle screen) {
this.setScale(atomContainer);
Rectangle2D bounds = BoundsCalculator.calculateBounds(atomContainer);
this.modelCenter = new Point2d(bounds.getCenterX(), bounds.getCenterY());
this.drawCenter = new Point2d(screen.getCenterX(), screen.getCenterY());
this.setup();
}
So the setup function does some stuff but what interests me are the two lines with the modelCenter and drawCenter that get the dimensions from the bounding box and the screen, respectively. What if I manually change the drawCenter? Can I effectively translate the molecule image within a larger image? I trackdown setDrawCenter and add the following lines AFTER the setup function.
(-> renderer (.setDrawCenter 200 200))
Bang! This does the trick. I can now use large screen image but center the molecule anywhere in that larger screen. Following the hint from the mailing list, I now look for a zoom method and find it, allowing me to set the zoom. With rendering, rotation, zooms and scaling I should be able to generate a lot of types of images. If I want to display multiple images I’ll need to modify the original function to this:
;helper function to allow passing of values
(defn mol [atomcontainer & {:keys [zoom x y ]
:or {x 0 y 0 zoom 1}}]
{:mol atomcontainer :x x :y y :zoom zoom})
;updated funciton can zoom and translate
(defn simplerender [mols]
(let [gen (doto (ArrayList.)
(.add (new BasicSceneGenerator))
(.add (new StandardGenerator font)))
image (BufferedImage. 800 300 1)
drawArea (Rectangle. 800 300)
g (doto (.getGraphics image) (.fillRect 0 0 800 300))
renderer (AtomContainerRenderer. gen (new AWTFontManager))]
(doseq [mol mols]
(-> renderer (.setZoom (:zoom mol)))
(-> renderer (. setup (:mol mol) drawArea))
(-> renderer (.setDrawCenter (:x mol) (:y mol)))
(-> renderer (. paint (:mol mol) (new AWTDrawVisitor g))))
image))
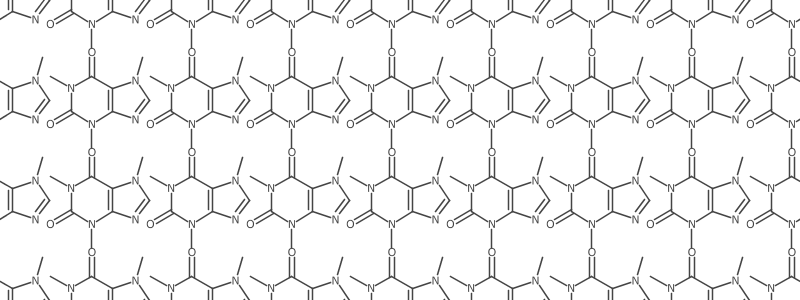
Using those two functions (the modified original, and helper that lets me pass x, y, and zoom) I can take molecule and generate a bunch of coordinates, and plot all of the translated molecules on the same canvas. The code below makes the repetitive caffeine pic at the header of this post.
;generate caffeine molecules at lots of positions
(def mols (for [x (range 0 900 100)
y (range 0 900 100)] (mol caffeine :x x :y y :zoom 0.5)))
;draw them to the same image and save it
(save (simplerender mols ))
And a variation on a theme that will generate the same caffeine molecules but on seperate images. Then make them into a gif.
(def molimages
(for [y (range 0 300 100)
x (range 0 900 100)]
(simplerender [(mol caffeine :x x :y y :zoom 0.5)])))
(write-gif "/Users/zachpowers/Desktop/caffeine_grid_anim.gif" molimages :delay 10 :delaylast 10)

Take Home.
I am interested in using CDK to make animations of chemical structures. CDK provides a fantastic tool for generating high quality structures. But what if you want to make your own scenes that have chemical structures as part of them? Then you need to be able to place the molecules in the correct orientation and positions. Here I’ve shown a that a basic understanding of the graphing tools allow you to specify some of these properties and create scenes, or animateable scenes from chemical structures.So far I discovered the ability to use GeometryTools/rotate for rotation, and the ability to set the zoom and center for an AtomContainer Renderer.
There is still a lot more to know - for example can you find the EXACT position of particular atoms on the screen? Once you can control this level of detail you can simulate bond formations/breakages and can layout biologically meaningful animations. If I can master a bit more of the CDK depiction library it shouldn’t be too difficult to make a Clojure wrapper that facilitates fine-grained image generation. Thanks again to the CDK team.