Simons Microbiome Data Analysis MiniConference
Heres a few thoughts/notes on April 1-2 miniconference organized by the Simons Foundation ([conference link][]). This conference is primarily attended by statisticians,and tool developers seeking to improve the software and analytical choices used by microbiome scientists to analyze data. Consequently, the talks were primarily focused on methods and software as opposed to, say, biological findings. Given my background I was particuarly interested in the section an analysis of microbial metabolites and the section on the detection and elimination of bias & error.
Highlights#
Peptide Natural Products#
In the metabolite detection session the most interesting talk was presented by Dr. Hosein Mohimani, a former postdoc in the Dorrestein Lab who is now at his own lab at Carnegie Mellon. As befits the Dorrestein lineage, Hosein is interested in developing methods to detect metabolites of interest from MS/MS spectra. His talk centred on the development of a method, and subsequent software tools, for detecting cyclic peptides from MS-MS spectra. One tool, Cyclonovo, developed along with Behar Behsaz, is specifically designed to detect fragmentation patterns characteristic of cyclic peptides. Two applictions of the tool were presented: 1) denovo discovery of cyclic peptide fragments from public MS/MS data sets (GNPS was mentioned numerous times) and 2) coupling of MS/MS data with the biosynthetic predictions derived from Antismash. In this way he can build a set of expectation-masses from the Adenylation-domain predictions in Antismash, and create an MS-fragment database which can be used to query MS-spectra.
Befitting the setting, Hosein spent a great deal of time discussing how he encodes the expected fragmentation patterns (a row of 2000 binaries representing masses from 0 to 2000), and how this is queried against the MS/MS data (use matrix multiplication and scoring). As a result he also discussed some of the limitations - namely that every single modification to a peptide - e.g. fatty acid chain addition, halogenation, oxidation - will result in a completely different set of reference spectra that would need to be encoded. He has some ability to accomdate changes but it is currently limited to a single difference. As his interest is the application of these detection to billions of MS/MS spectra available in public datasets, the size of this reference set quickly becomes a computational challenge. He is investigating several dynamic programming approaches that allow him to dynamically identify spectra of interest which get more extensively searched.
After speaking with him and Behar Behsaz ( a current postdoc in the Dorrestein lab, and also a conference speaker who spoke about related methods) and learning a bit about the utility of his tools, I think it would be worthwhile looking into several of hits peptide-natural-product based tools:
- Dereplicator, Dereplicator+, Varquest, Cyclonovo, MetaMiner and
- NPDTools The distribution that seems to hold most of the tools above.
Bias Detection#
There was an excellent talk by Benjamin Callahan, the author of the DADA2 R package. His talk was an attempt to step back from microbiome analyses to assess the sources of bias in a comprehensive way and to lay out some strategies for addressing bias. His talk was based on an available preprint, ([bias preprint][https://www.biorxiv.org/content/10.1101/559831v1]). There were two major ideas under investigation:
- observational bias creep in at EVERY step of the protocol.
- since sequencing counts only contains information on the relative abundances of taxa/ marker genes, if bias is not taken into account, relativ changes can be completely wrong.
These two are indicated by the two figures in his paper.
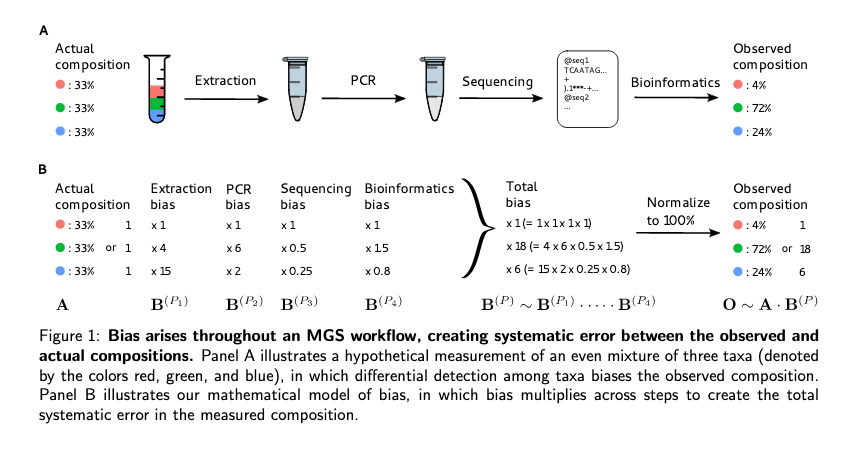 sources of observational bias
sources of observational bias
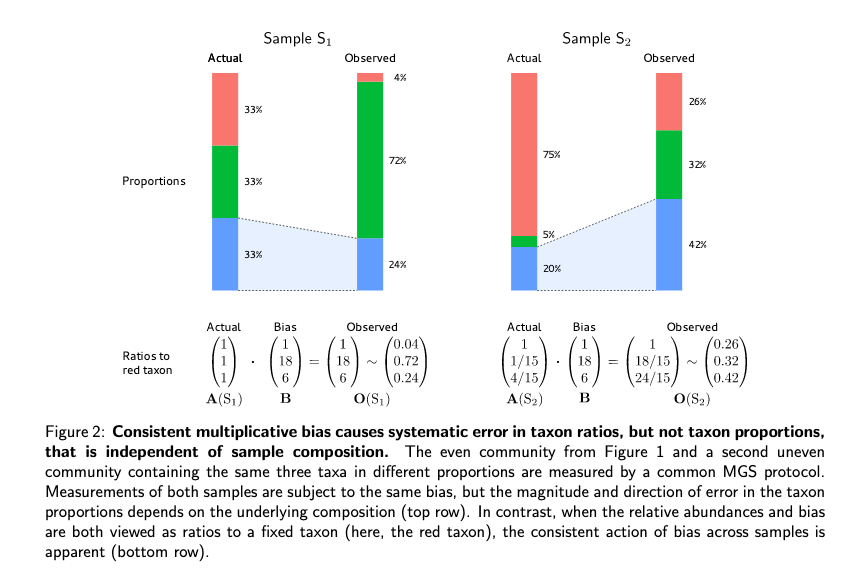 importance of composition in calculating relative abundances
importance of composition in calculating relative abundances
His talk centered on efforts to see if a model could be developed to correct for biases and he drew upon data from an earlier experimental study on bias where a set of controlled mixture of initial samples were processed and the outputs compared with the inputs. Ben and his colleagues were able to derived a correction factor that could be applied to the abundance data to correctly account for the changes in relative abundance.
This talk was a beautiful application of simple statistical thinking to a pervasive problem in microbiome science. Ben laid out a path forward which would include changing how experimental controls are designed to make sure that a bias correction measure can be included. The main idea is that the sources of bias will be constant in each sample so if we normalize to a common component of hte sample our relative abundances should be the same (i.e. we can correct for the biases relative to a control).
Relatedly, David Clausen, a student in Amy Willis’ group, presented their analysis of the Microbiome Quality Control (MBQC) study in the context of classification of compositions.
The picture he painted looked bleak: models on data generated in one lab could not be used to test models from a different lab - even though the samples were identical (just processed independently). No solutions (such as batch correction) were proposed however, so it’s not clear if the classification task is unrecoverable without explicit specification of all the errors (such as suggested by Ben Callahan).
Source Contamination#
Liat Shenhav presented FEAST, a method for tracking the source of microbial mixtures. This is, in principal, similar to SourceTracker but the model allows for the specification of unknown source pool.
It uses an EM algorithm to solve the objective, and so is much faster than Bayesian methods and scales to thousands of possible population ‘sinks’. Something like this might be used to deconvolute pools with mixed populations of contaminants, though it is not clear if the procedure guarantees identifiability or convergence.
P. Janganathan, a Postdococtoral Fellow at Standford from Susan Holmes’ lab, gave a talk about a method for detecting and removing bias and contamination from micribiome samples called BARBI
BARBI models count distributions as mixture models and uses a Bayesian method to tease apart the convoluted signals. However, it was not clear to whether this method is scalable for very large OTU/ very sparse table OTU data of the sort we encounter in Natural product sequence profiling.
Other Highlights#
Effect sizes#
Greg Gloor presented work that advocated for using “effect sizes” instead of p-values. For example, when looking at differential taxa under and experimental perturbation. Carefully constructed effect size estimates seem to be more robust to false positives, even in underpowered study designs.
When the counts data is from metagenomic sequencing, he made an interesting observation that feature detection based on p-value cutoffs are more sensitive than effect sizes in the ‘sample-depth’ vs ‘sample-size’ tradeoff (i.e. multiplexing doesn’t matter so much when you resample low abundant genes).
CAMI#
The CAMI team presented its new challenge and there are several tools worth investigating that deal with phylogeny and taxonomy. It would be interesting to checkout their benchmarking results to see if there are any (metagnome assembly) tools that are better for recovering BGCs.
Protein structure and annotation#
Vladimir Gligorijevic of the Flatiron institute was using Deep Learning (CNN and graph-CNNs) to predict GO terms from primary protein sequence.
The use of Deep neural networks to automate higher-order features is interesting, especially since it does so much better than sequence homology or even kmer methods. But it’s quite possible that this early iteration of the tool is overkill (compared to HMMs or shallower networks).
The graph-CNN methods requires structure prediction with Rosetta first, so this is especially computationally expensive.
mmvec#
Jamie Morton presented his work on MMvec, a word2-vec like process for training microbial data and metabolite data to find positive associations between OTU counts and metabolite abundances.
He showed some work on a lung microbiome study that had microbiome data AND metabolomic data. Primarily he could detect metabolites associated with P.Aeuriginosa, but this is mostly because those were well annotated metabolites.